Sakai, T., Martinez-Anaya, C., Contreras, M. P., Kamoun, S., Wu, C.-H., & Adachi, H. (2023). The NRC0 gene cluster of sensor and helper NLR immune receptors is functionally conserved across asterid plants. BioRxiv. https://doi.org/10.1101/2023.10.23.563533
Abstract
NLR (nucleotide-binding domain and leucine-rich repeat-containing) proteins can form complex receptor networks to confer innate immunity. NRCs are phylogenetically related nodes that function downstream of a massively expanded network of disease resistance proteins that protect against multiple plant pathogens. Here, we used phylogenomic methods to reconstruct the macroevolution of the NRC family. One of the NRCs, we termed NRC0, is the only family member shared across asterid plants, leading us to investigate its evolutionary history and genetic organization. In several asterid species, NRC0 is genetically clustered to other NLRs that are phylogenetically related to NRC-dependent disease resistance genes. This prompted us to hypothesize that the ancestral state of the NRC network is an NLR helper-sensor gene cluster that was present early during asterid evolution. We validated this hypothesis by demonstrating that NRC0 is essential for the hypersensitive cell death induced by its genetically linked sensor NLR partners in four divergent asterid species: tomato, wild sweet potato, coffee and carrot. In addition, activation of a sensor NLR leads to high-order complex formation of its genetically linked NRC0 similar to other NRCs. Our findings map out contrasting evolutionary dynamics in the macroevolution of the NRC network over the last 125 million years from a functionally conserved NLR gene cluster to a massive genetically dispersed network.
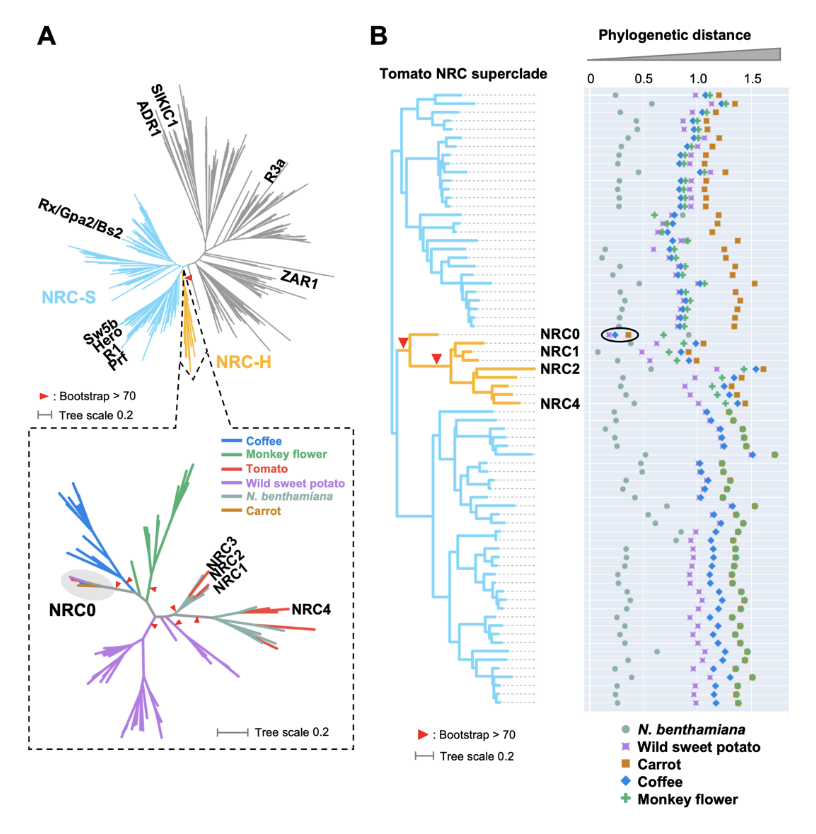
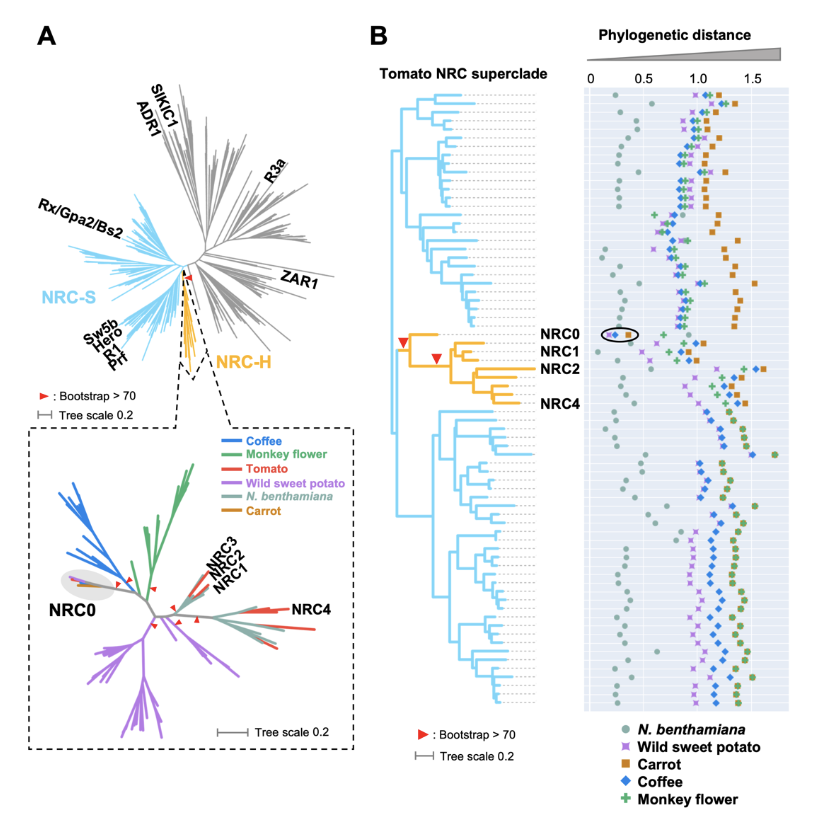
NRC0 is the most conserved helper clade NRC in asterids.(A) Phylogeny of NLRs identified from asterids (carrot, monkey flower, coffee, wild sweet potato, Nicotiana benthamiana, and tomato). The maximum likelihood phylogenetic tree was generated in RAxML version 8.2.12 with JTT model using NB-ARC domain sequences of 1,661 NLRs identified from carrot, monkey flower, coffee, wild sweet potato, N. benthamiana, and tomato reference genome by using the NLRtracker and 39 functionally validated NLRs. In the top left phylogenetic tree, the NRC superclades are described with different branch color codes: NRC-helper (NRC-H) subclade shown in orange and NRC-sensor (NRC-S) subclades shown in light blue. The bottom left phylogenetic tree describes NRC-H subclade with different color codes based on plant species. Red arrow heads indicate bootstrap support > 0.7 and is shown for the relevant nodes. (B) Phylogenetic distance of two NRC-H and NRC-S nodes between tomato and other plant species. The phylogenetic distance was calculated from the NB-ARC phylogenetic tree shown in A. The closest distances are plotted with different colors based on plant species. Representative tomato NRC-H are highlighted.
Adachi, H., Sakai, T., Kourelis, J., Pai, H., Gonzalez Hernandez, J. L., Utsumi, Y., Seki, M., Maqbool, A., & Kamoun, S. (2023). Jurassic NLR: Conserved and dynamic evolutionary features of the atypically ancient immune receptor ZAR1.The Plant Cell, 35(10), 3662–3685. https://doi.org/10.1093/plcell/koad175
Abstract
Plant nucleotide-binding leucine-rich repeat (NLR) immune receptors generally exhibit hallmarks of rapid evolution, even at the intraspecific level. We used iterative sequence similarity searches coupled with phylogenetic analyses to reconstruct the evolutionary history of HOPZ-ACTIVATED RESISTANCE1 (ZAR1), an atypically conserved NLR that traces its origin to early flowering plant lineages ∼220 to 150 million yrs ago (Jurassic period). We discovered 120 ZAR1 orthologs in 88 species, including the monocot Colocasia esculenta, the magnoliid Cinnamomum micranthum, and most eudicots, notably the Ranunculales species Aquilegia coerulea, which is outside the core eudicots. Ortholog sequence analyses revealed highly conserved features of ZAR1, including regions for pathogen effector recognition and cell death activation. We functionally reconstructed the cell death activity of ZAR1 and its partner receptor-like cytoplasmic kinase (RLCK) from distantly related plant species, experimentally validating the hypothesis that ZAR1 evolved to partner with RLCKs early in its evolution. In addition, ZAR1 acquired novel molecular features. In cassava (Manihot esculenta) and cotton (Gossypium spp.), ZAR1 carries a C-terminal thioredoxin-like domain, and in several taxa, ZAR1 duplicated into 2 paralog families, which underwent distinct evolutionary paths. ZAR1 stands out among angiosperm NLR genes for having experienced relatively limited duplication and expansion throughout its deep evolutionary history. Nonetheless, ZAR1 also gave rise to noncanonical NLRs with integrated domains and degenerated molecular features.
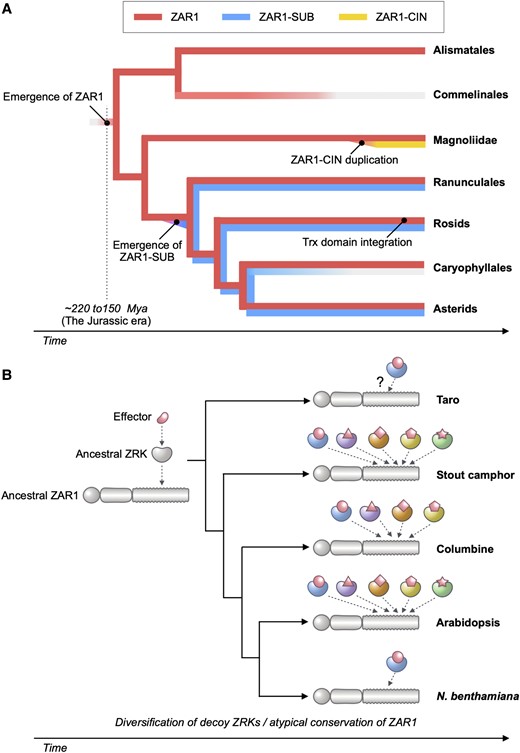
Coevolution of ZAR1 and ZRK genes in angiosperms. A) We propose that the ancestral ZAR1 gene has emerged ∼220 to 150 million yrs ago (Mya) before monocot and eudicot lineages split. ZAR1 is a widely conserved CC-NLR in angiosperms, but it is likely that ZAR1 was lost in the monocot lineage, Commelinales. A sister clade paralog ZAR1-SUB has emerged early in the eudicot lineages and may have been lost in Caryophyllales. Another sister clade paralog ZAR1-CIN was duplicated from the ZAR1 gene and expanded in the Magnoliidae C. micranthum. Trx domain integration to C terminus of ZAR1 has independently occurred in few rosid lineages. B)ZAR1 has coevolved with partner ZRK gene for pathogen effector recognition since the Jurassic era. During the coevolution, ZRKs diversified to catch up with fast-evolving effectors.
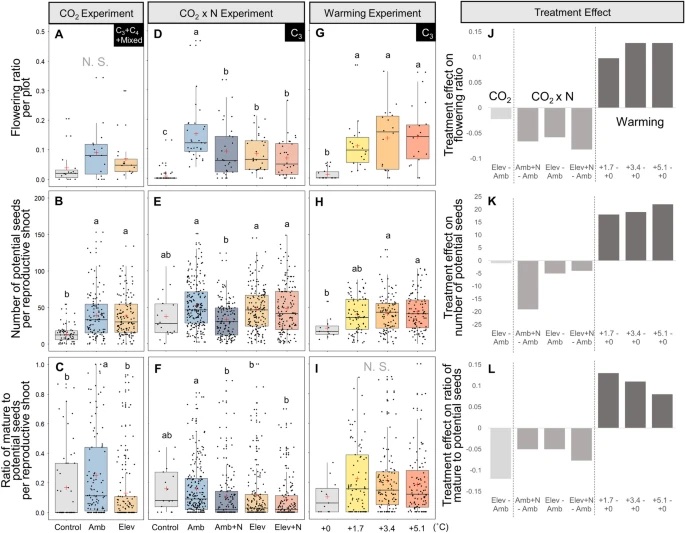
The expansion of many wetland species is a function of both clonal propagation and sexual reproduction. The production of ramets through clonal propagation enables plants to move and occupy space near parent ramets, while seeds produced by sexual reproduction enable species to disperse and colonize open or disturbed sites both near and far from parents. The balance between clonal propagation and sexual reproduction is known to vary with plant density but few studies have focused on reproductive allocation with density changes in response to global climate change. Schoenoplectus americanus is a widespread clonal wetland species in North America and a dominant species in Chesapeake Bay brackish tidal wetlands. Long-term experiments on responses of S. americanus to global change provided the opportunity to compare the two modes of propagation under different treatments. Seed production increased with increasing shoot density, supporting the hypothesis that factors causing increased clonal reproduction (e.g., higher shoot density) stimulate sexual reproduction and dispersal of genets. The increase in allocation to sexual reproduction was mainly the result of an increase in the number of ramets that flowered and not an increase in the number of seeds per reproductive shoot, or the ratio between the number of flowers produced per inflorescence and the number of flowers that developed into seeds. Seed production increased in response to increasing temperatures and decreased or did not change in response to increased CO2 or nitrogen. Results from this comparative study demonstrate that plant responses to global change treatments affect resource allocation and can alter the ability of species to produce seeds.
Sugihara, Y., Abe, Y., Takagi, H., Abe, A., Shimizu, M., Ito, K., Kanzaki, E., Oikawa, K., Kourelis, J., Langner, T., Win, J., Białas, A., Lüdke, D., Contreras, M. P., Chuma, I., Saitoh, H., Kobayashi, M., Zheng, S., Tosa, Y., Banfield, M., Kamoun, S., Terauchi, R., & Fujisaki, K. (2023). Disentangling the complex gene interaction networks between rice and the blast fungus identifies a new pathogen effector. PLOS Biology, 21(1), e3001945-. https://doi.org/10.1371/journal.pbio.3001945
Abstract
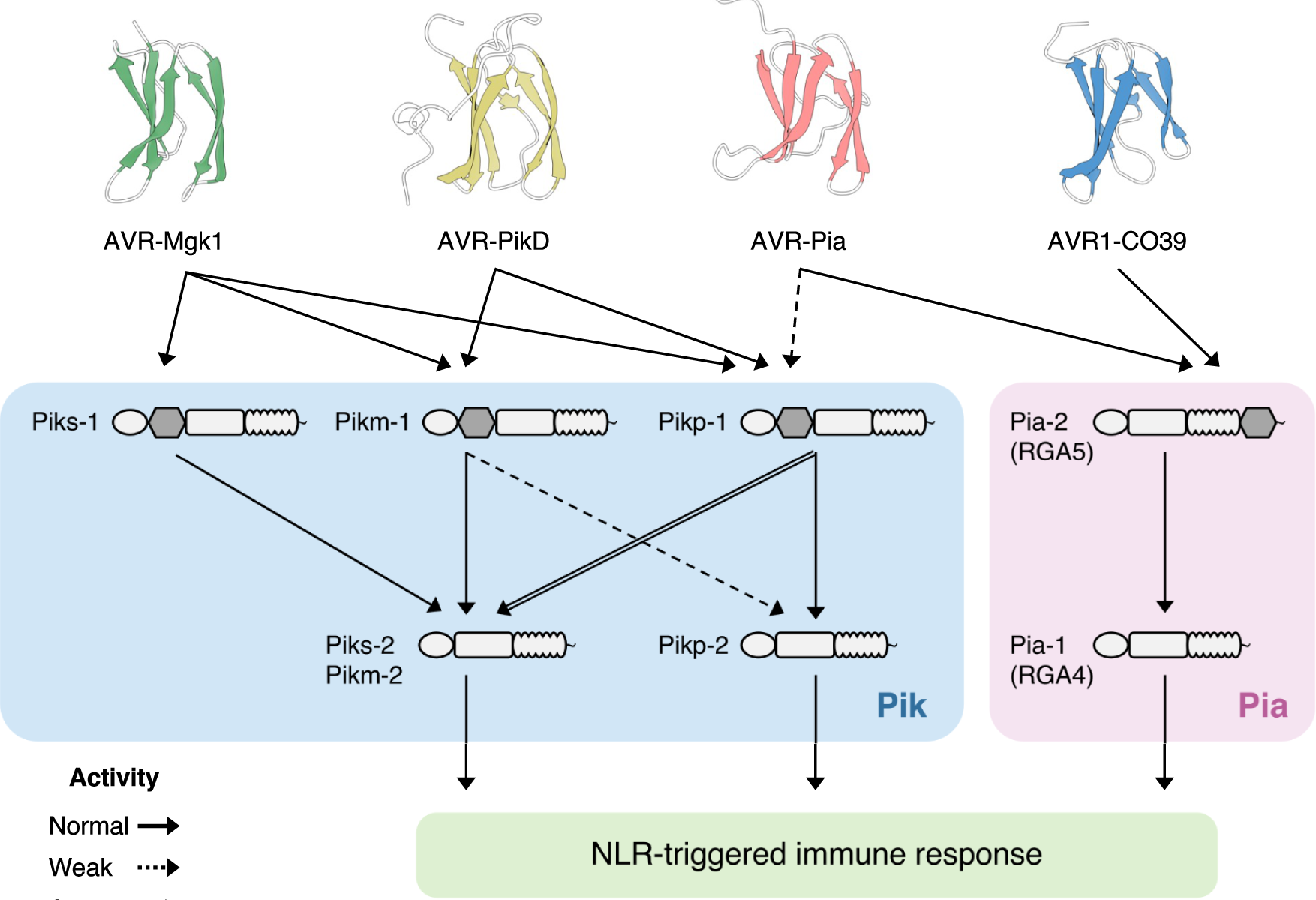
Studies focused solely on single organisms can fail to identify the networks underlying host–pathogen gene-for-gene interactions. Here, we integrate genetic analyses of rice (Oryza sativa, host) and rice blast fungus (Magnaporthe oryzae, pathogen) and uncover a new pathogen recognition specificity of the rice nucleotide-binding domain and leucine-rich repeat protein (NLR) immune receptor Pik, which mediates resistance to M. oryzae expressing the avirulence effector gene AVR-Pik. Rice Piks-1, encoded by an allele of Pik-1, recognizes a previously unidentified effector encoded by the M. oryzae avirulence gene AVR-Mgk1, which is found on a mini-chromosome. AVR-Mgk1 has no sequence similarity to known AVR-Pik effectors and is prone to deletion from the mini-chromosome mediated by repeated Inago2 retrotransposon sequences. AVR-Mgk1 is detected by Piks-1 and by other Pik-1 alleles known to recognize AVR-Pik effectors; recognition is mediated by AVR-Mgk1 binding to the integrated heavy metal-associated (HMA) domain of Piks-1 and other Pik-1 alleles. Our findings highlight how complex gene-for-gene interaction networks can be disentangled by applying forward genetics approaches simultaneously to the host and pathogen. We demonstrate dynamic coevolution between an NLR integrated domain and multiple families of effector proteins.
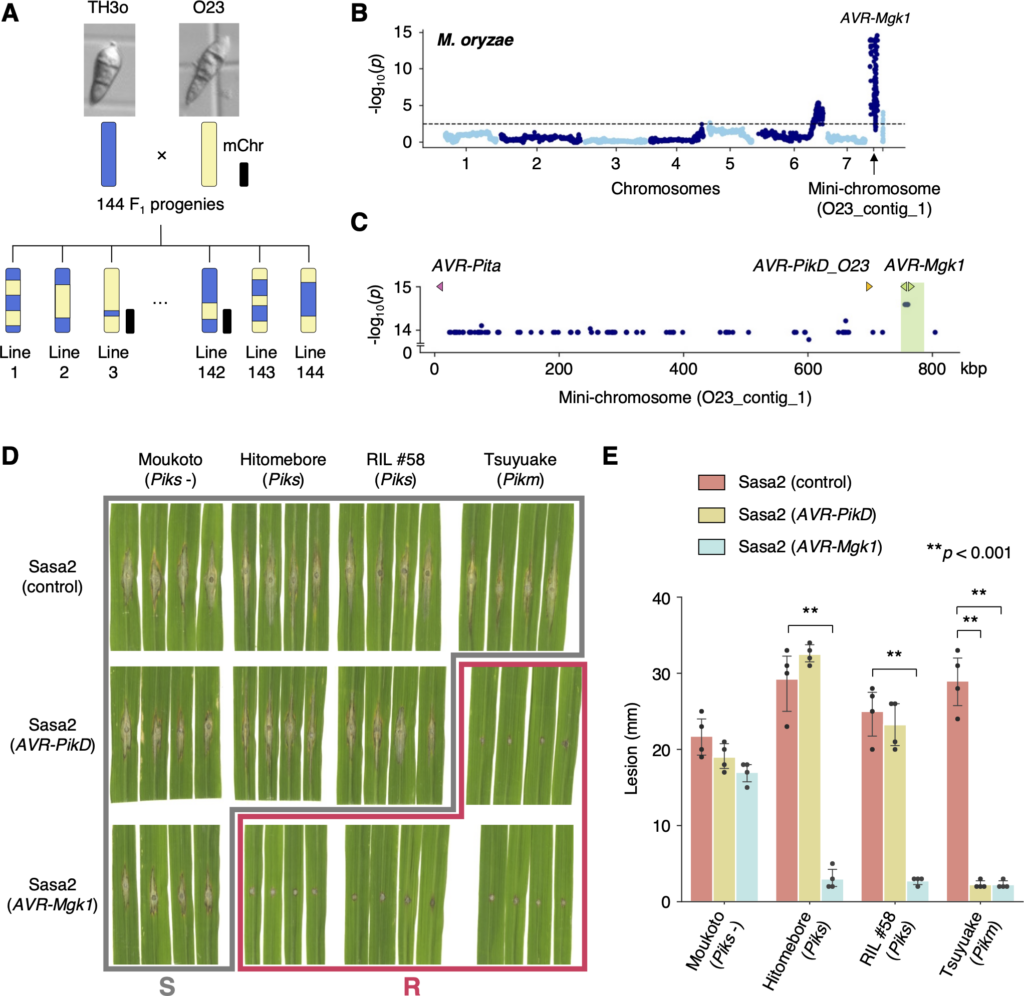
Fig. M. oryzae genetic analysis identifies an AVR gene, AVR-Mgk1, encoded on a mini-chromosome. (A) Schematic representations of the F1 progeny generated after a cross between M. oryzae isolates TH3o and O23. We subjected all F1 progeny to whole-genome sequencing. O23 possesses a mini-chromosome. (B) Genetic association of the TH3o × O23 F1 progeny using infection lesion size on RIL #58 (Pish -, Piks +) rice plants as a trait. The vertical axis indicates -log10(p), where the p-value is how likely the marker shows association with a trait due to random chance. The dashed line shows the p-value corresponding to a false discovery rate of 0.05. The association analysis based on the O23 reference genome identified AVR-Mgk1, encoded on the mini-chromosome sequence O23_contig_1, as an AVR gene. O23_contig_1 was not present in the TH3o genome and was unique to the O23 genome. We used 7,867 SNP markers for chromosomes 1–7 and 265 presence/absence markers for the other contigs. (C) p-values for O23_contig_1 with annotated AVRs. We also detected AVR-Pita and AVR-PikD in O23_contig_1. AVR-PikD in O23_contig_1 contains a frameshift mutation, so we named this variant AVR-PikD_O23. The region encoding 2 AVR-Mgk1 genes and showing lower p-values is highlighted in green. Nucleotide sequences of the 2 AVR-Mgk1 genes, arranged in a head-to-head orientation, are identical. (D) Results of punch inoculation assays using M. oryzae isolate Sasa2 transformed with AVR-PikD or AVR-Mgk1. Wild-type Sasa2 infected all the cultivars tested in this study. The Sasa2 transformant expressing AVR-PikD infected RIL #58 (Piks), but that expressing AVR-Mgk1 did not infect RIL #58 (Piks) or Tsuyuake (Pikm) rice plants. (E) Quantification of the lesion size in (D). Asterisks indicate statistically significant differences (p < 0.001, two-sided Welch’s t test). The data underlying Fig 4B and 4C and 4E can be found in S1 Data. AVR, avirulence; RIL, recombinant inbred line; SNP, single nucleotide polymorphism. https://doi.org/10.1371/journal.pbio.3001945.g004