
研究室のメンバー一覧を更新しました。
The NRC0 gene cluster of sensor and helper NLR immune receptors is functionally conserved across asterid plants
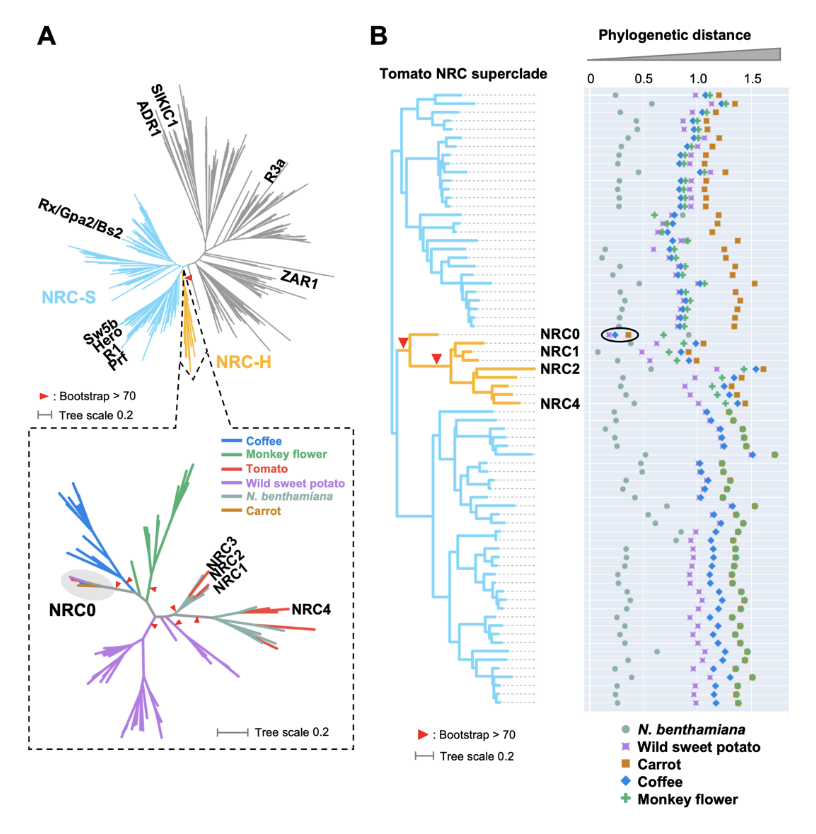
New paper was posted at bioRxiv
Sakai, T., Martinez-Anaya, C., Contreras, M. P., Kamoun, S., Wu, C.-H., & Adachi, H. (2023). The NRC0 gene cluster of sensor and helper NLR immune receptors is functionally conserved across asterid plants. BioRxiv. https://doi.org/10.1101/2023.10.23.563533
Abstract
NLR (nucleotide-binding domain and leucine-rich repeat-containing) proteins can form complex receptor networks to confer innate immunity. NRCs are phylogenetically related nodes that function downstream of a massively expanded network of disease resistance proteins that protect against multiple plant pathogens. Here, we used phylogenomic methods to reconstruct the macroevolution of the NRC family. One of the NRCs, we termed NRC0, is the only family member shared across asterid plants, leading us to investigate its evolutionary history and genetic organization. In several asterid species, NRC0 is genetically clustered to other NLRs that are phylogenetically related to NRC-dependent disease resistance genes. This prompted us to hypothesize that the ancestral state of the NRC network is an NLR helper-sensor gene cluster that was present early during asterid evolution. We validated this hypothesis by demonstrating that NRC0 is essential for the hypersensitive cell death induced by its genetically linked sensor NLR partners in four divergent asterid species: tomato, wild sweet potato, coffee and carrot. In addition, activation of a sensor NLR leads to high-order complex formation of its genetically linked NRC0 similar to other NRCs. Our findings map out contrasting evolutionary dynamics in the macroevolution of the NRC network over the last 125 million years from a functionally conserved NLR gene cluster to a massive genetically dispersed network.
Jurassic NLR: Conserved and dynamic evolutionary features of the atypically ancient immune receptor ZAR1
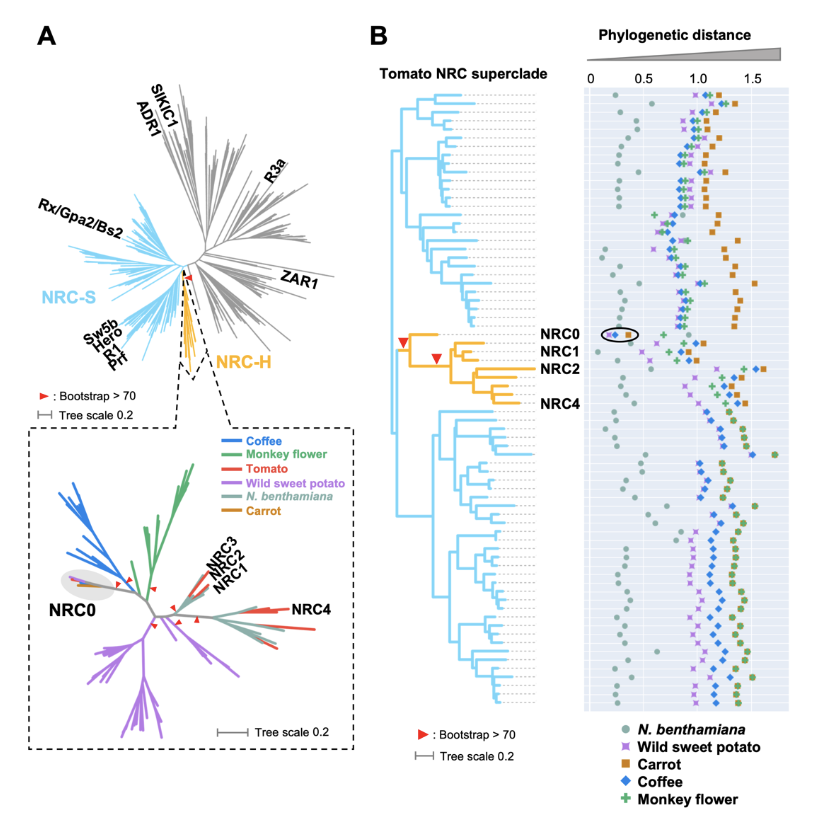
New paper from our laboratory
Adachi, H., Sakai, T., Kourelis, J., Pai, H., Gonzalez Hernandez, J. L., Utsumi, Y., Seki, M., Maqbool, A., & Kamoun, S. (2023). Jurassic NLR: Conserved and dynamic evolutionary features of the atypically ancient immune receptor ZAR1. The Plant Cell, 35(10), 3662–3685. https://doi.org/10.1093/plcell/koad175
Abstract
Plant nucleotide-binding leucine-rich repeat (NLR) immune receptors generally exhibit hallmarks of rapid evolution, even at the intraspecific level. We used iterative sequence similarity searches coupled with phylogenetic analyses to reconstruct the evolutionary history of HOPZ-ACTIVATED RESISTANCE1 (ZAR1), an atypically conserved NLR that traces its origin to early flowering plant lineages ∼220 to 150 million yrs ago (Jurassic period). We discovered 120 ZAR1 orthologs in 88 species, including the monocot Colocasia esculenta, the magnoliid Cinnamomum micranthum, and most eudicots, notably the Ranunculales species Aquilegia coerulea, which is outside the core eudicots. Ortholog sequence analyses revealed highly conserved features of ZAR1, including regions for pathogen effector recognition and cell death activation. We functionally reconstructed the cell death activity of ZAR1 and its partner receptor-like cytoplasmic kinase (RLCK) from distantly related plant species, experimentally validating the hypothesis that ZAR1 evolved to partner with RLCKs early in its evolution. In addition, ZAR1 acquired novel molecular features. In cassava (Manihot esculenta) and cotton (Gossypium spp.), ZAR1 carries a C-terminal thioredoxin-like domain, and in several taxa, ZAR1 duplicated into 2 paralog families, which underwent distinct evolutionary paths. ZAR1 stands out among angiosperm NLR genes for having experienced relatively limited duplication and expansion throughout its deep evolutionary history. Nonetheless, ZAR1 also gave rise to noncanonical NLRs with integrated domains and degenerated molecular features.
シンポジウムと展示の開催のお知らせ

国際シンポジウム
「コムギ研究の新展開:100年の研究史を反映した生物遺伝資源とゲノミクス、そして未来へ」
日 時:10月13日(金)9:00-
場 所:京都大学吉田キャンパス北部構内 北部総合教育研究等1階 益川記念ホール

紹介展示
「田中正武アーカイブズとNBRP・コムギリソース」
日 時:10月12日(木)- 14日(土)
場 所:京都大学吉田キャンパス北部構内 旧演習林事務室ラウンジ
Reproductive Responses to Increased Shoot Density and Global Change Drivers in a Widespread Clonal Wetland Species, Schoenoplectus americanus
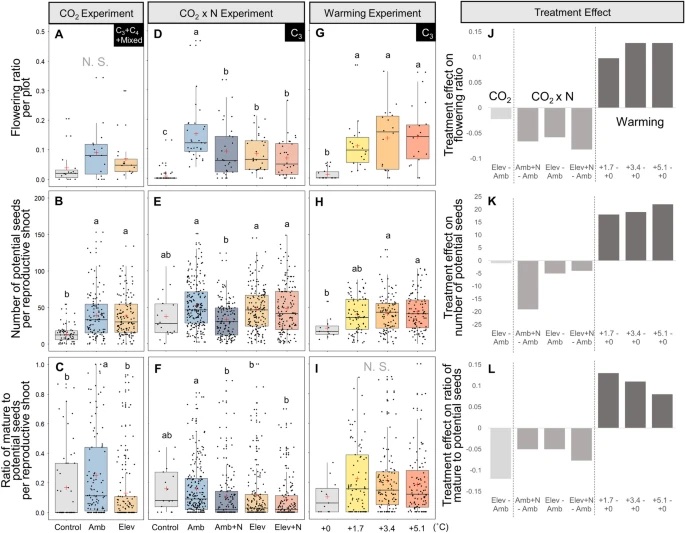
New paper from our laboratory
Kudoh, A., Megonigal, J. P., Langley, J. A., Noyce, G. L., Sakai, T., & Whigham, D. F. (2023). Reproductive Responses to Increased Shoot Density and Global Change Drivers in a Widespread Clonal Wetland Species, Schoenoplectus americanus. Estuaries and Coasts. https://doi.org/10.1007/s12237-023-01249-z
Abstract
The expansion of many wetland species is a function of both clonal propagation and sexual reproduction. The production of ramets through clonal propagation enables plants to move and occupy space near parent ramets, while seeds produced by sexual reproduction enable species to disperse and colonize open or disturbed sites both near and far from parents. The balance between clonal propagation and sexual reproduction is known to vary with plant density but few studies have focused on reproductive allocation with density changes in response to global climate change. Schoenoplectus americanus is a widespread clonal wetland species in North America and a dominant species in Chesapeake Bay brackish tidal wetlands. Long-term experiments on responses of S. americanus to global change provided the opportunity to compare the two modes of propagation under different treatments. Seed production increased with increasing shoot density, supporting the hypothesis that factors causing increased clonal reproduction (e.g., higher shoot density) stimulate sexual reproduction and dispersal of genets. The increase in allocation to sexual reproduction was mainly the result of an increase in the number of ramets that flowered and not an increase in the number of seeds per reproductive shoot, or the ratio between the number of flowers produced per inflorescence and the number of flowers that developed into seeds. Seed production increased in response to increasing temperatures and decreased or did not change in response to increased CO2 or nitrogen. Results from this comparative study demonstrate that plant responses to global change treatments affect resource allocation and can alter the ability of species to produce seeds.